21 July 2014 by Remco Bouckaert
SNAPP treats each SNP as having its own gene tree, but what happens when there is data missing for some sites for a SNP? SNAPP simply assumes these taxa do not exist in the gene tree. If you have a species containing 3 lineages for which one has missing data, SNAPP assumes there is a gene tree with only 2 lineages for that species. So, what happens when all 3 lineages is missing? Previous versions of SNAPP (v1.1.5 and before) could not handle this situation and just removed these sites from the data. This can be a problem when you do species delimitation using BFD*; when you split a species and data is missing, some sites may be deleted that are not deleted when doing the un-split analysis. As a results, marginal likelihoods are calculated for different data sets, so these are not comparable. In v1.1.6, no sites with missing data are deleted any more, even when this leaves some species having zero lineages. SNAPP assumes the gene tree simply has no taxa in that species any more.
Another change in SNAPP v1.1.6 is that it uses the MCMC class from BEAST instead of SNAPP, which means there is no timeout and sampling from prior options any more, but this makes doing a path sampling analysis a lot easier.
First, make sure you have SNAPP at least v1.1.6 installed and Model Selection v1.0.2. Open the package manager (in BEAUti under the File/Manage packages menu) and select SNAPP, then click the Install button. After a little while a warning pops up that SNAPP is installed, and you might need to restart BEAUti to be able to set up a SNAPP analysis.

Then, select the SNAPP template, to set up a SNAPP analysis.

Click import data, and select a nexus file with your alignment. The taxa will automatically be assigned to a species by guessing based on the lineage names in the nexus files. You can change this of course.
In the Model Parameters panel, make sure you either calculate the values for u and v (see the Rough guide to SNAPP how to do this) or just click the estimate flag next to Mutation Rate U. When there is a sufficient amount of SNP data the estimates for U and V converge quite rapidly most of the time. Note: Leaving the default values for u and v and not estimating these mutation rates is almost surely going to lead to bad fits!
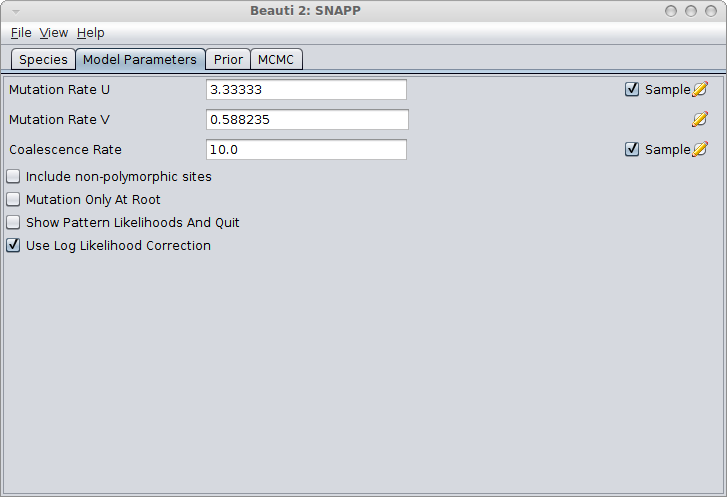
You can save this analysis as XML file, say runA.xml and run it with BEAST.
Now, if you want to calculate the marginal likelihood, you can start the AppStore application that is part of BEAST, and select the Path Sampler icon — either double click or click launch to start the Path sampler.
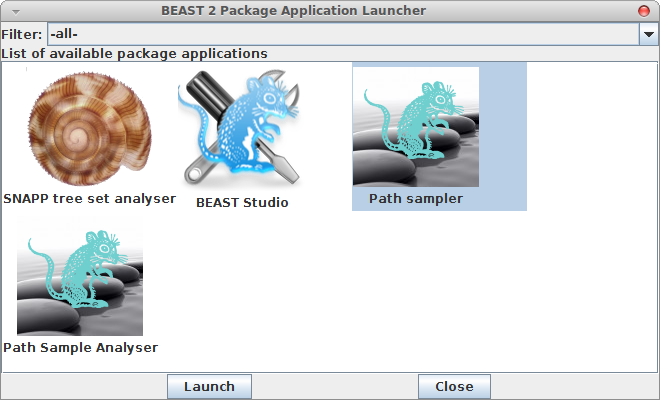
A window pops up where you can select the XML file you save from BEAUti and specify the parameters for the path sampling analysis.
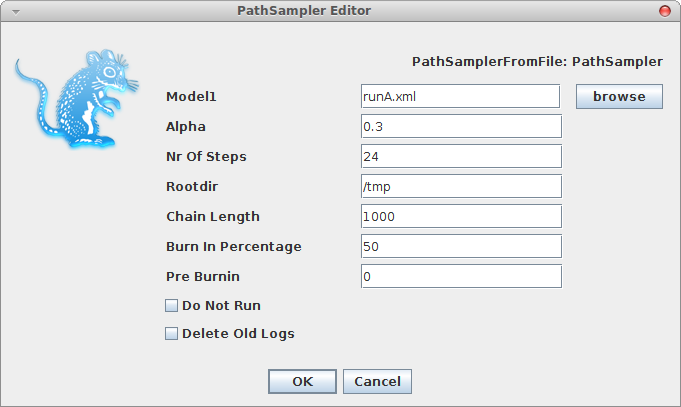
Once you click OK, the path sampling analysis is set up and run in a separate window, where after a little (or long — depending on the data) while the marginal likelihood will be printed.
