March 2016 by Remco Bouckaert, Tim Vaughan, Walter Xie, and Alexei Drummond
Recently, a few users reported problems with BEAST 2 performance, concluding it was worse than BEAST 1. This puzzled us, because BEAST 1 and 2 share the same core algorithms, and both spend most of their time doing phylogenetic likelihood calculations, which is optimised using BEAGLE, a library shared by both programs. In fact, recently we changed the way that BEAST 2 handles proportion invariant categories, saving some phylogenetic likelihood calculations, so in theory it should be faster when using a proportion of invariant sites in the model. So, we became curious whether there are real performance differences between BEAST 1 and 2 and decided to do a benchmark. We expected them to perform roughly the same on GTR and GRT+G analyses, and BEAST 2 to do better on GTR+I and GTR+G+I analyses.
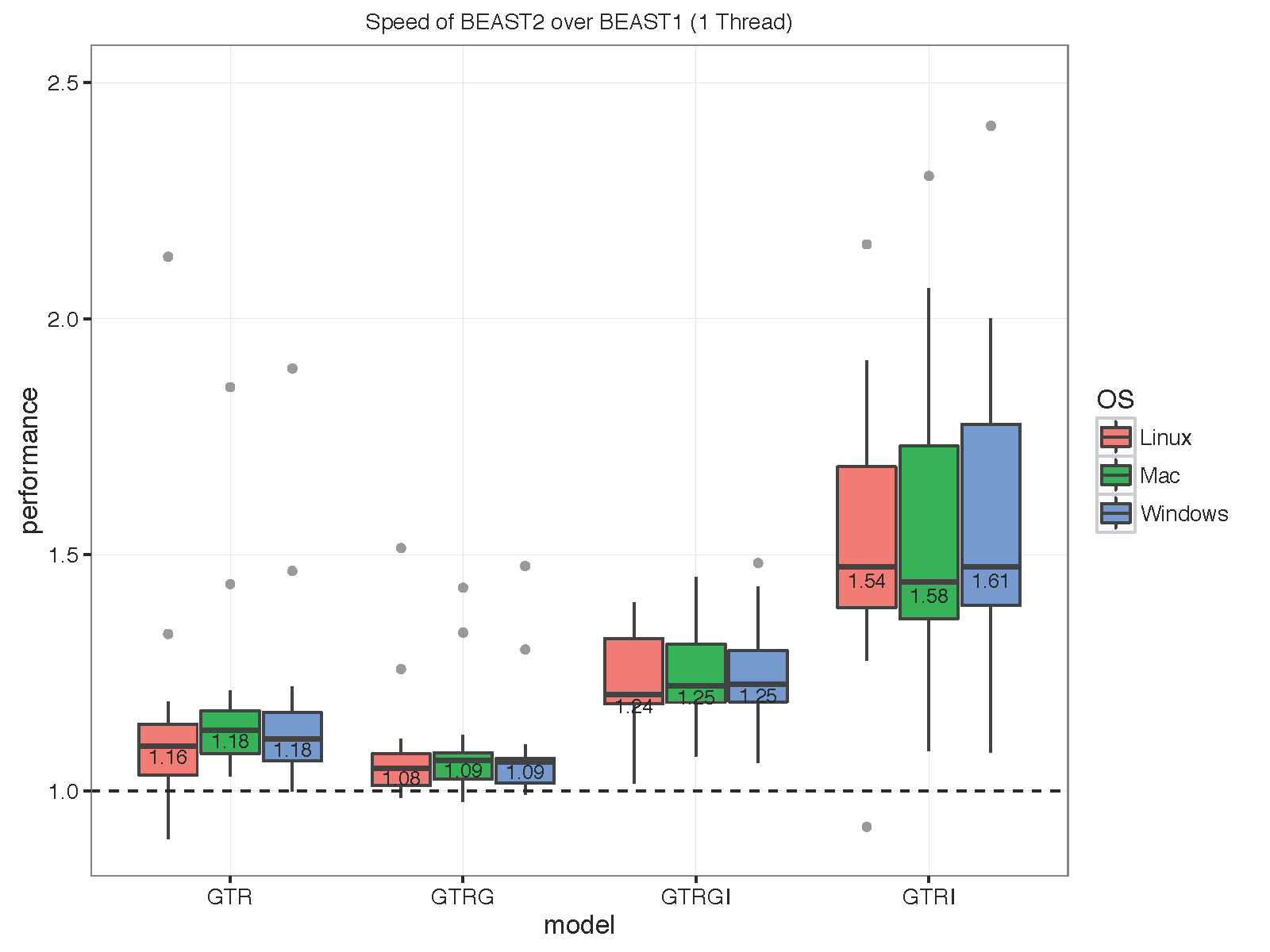
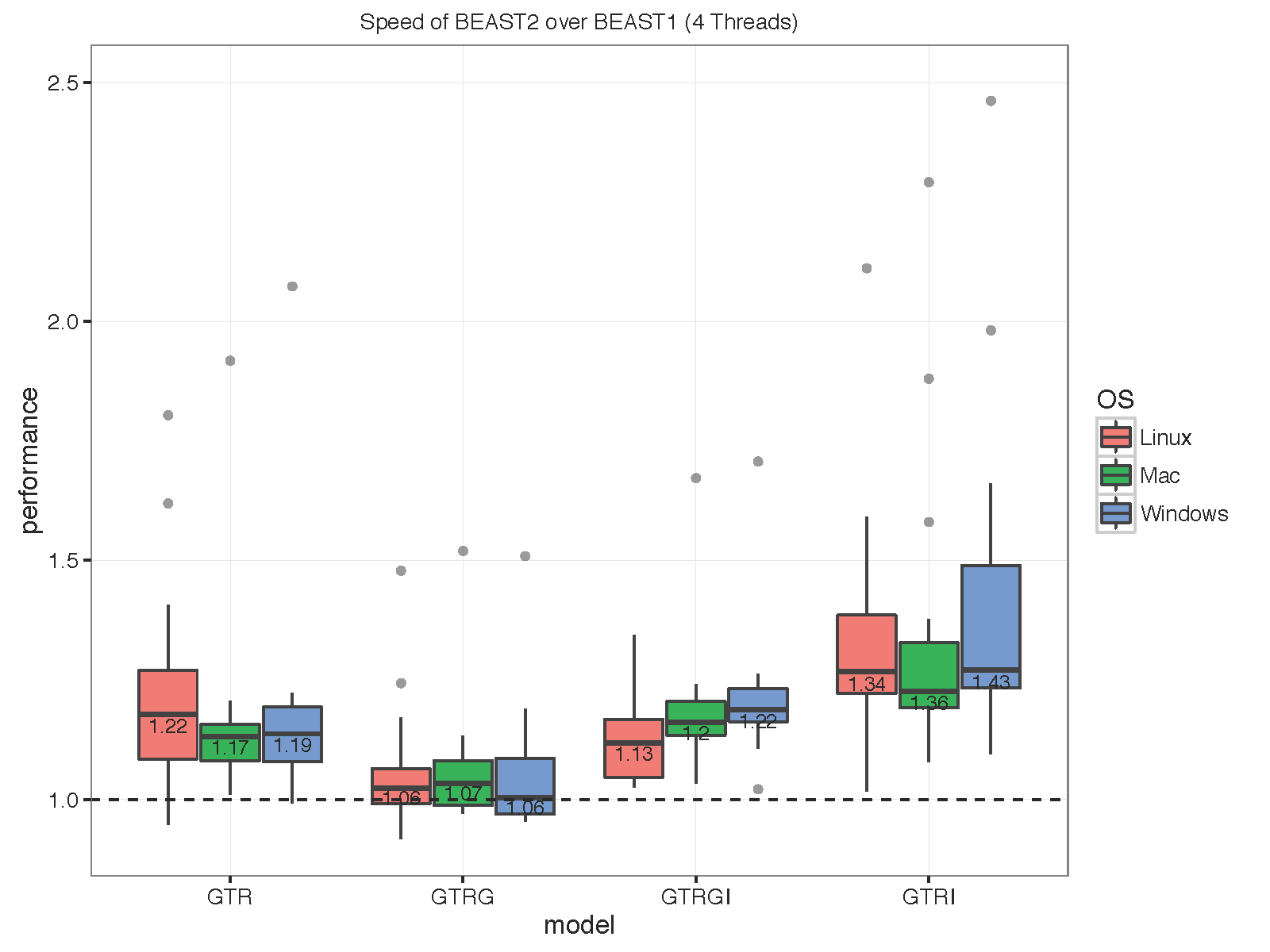
The picture below summarises the speed of BEAST 2 over BEAST 1 using 1, 2, 4 thread(s) in the 3 different operation systems. As you can see the performance is very similar for GTR and GTR+G, with BEAST2 being perhaps slight faster (although this could be due to debugging that BEAST1 performs at the start of the chain):
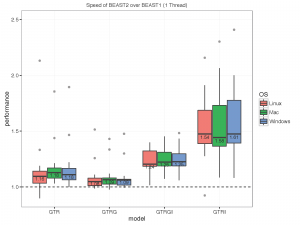
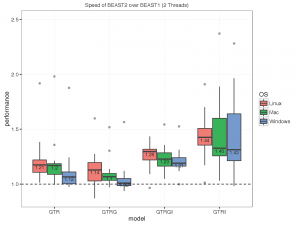
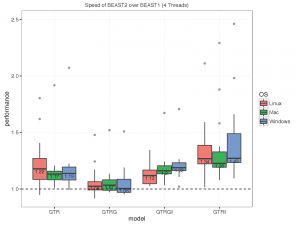
What we did
Analyses
BEAST can do many kinds of analyses, but for the purpose of this benchmark, we want to see whether the TreeLikelihood calculations, which typically dominate the computational time of MCMC runs, are comparable. To see the impact of the way BEAST 2 handles proportion invariant, we want to have an analysis with and without a proportion invariant category. And since many analyses use gamma rate heterogeneity with and without proportion invariants, we end up with four variants:
- GTR
- GTR + 4 gamma categories
- GTR + proportion invariant
- GTR + 4 gamma categories + proportion invariant
To keep things otherwise simple, we use a Yule tree prior, a strict clock and start with a random tree. To be practical, we set up the analysis in BEAUti 1 and 2, just importing an alignment, choosing the site model, setting the tree prior in BEAST 1 (BEAST 2 uses Yule by default) and save to file. As it turns out, the analyses produced that way are almost the same, but there are some small differences in the operator settings. Due to auto-optimisation, they will eventually become almost the same, but to make the two analyses as equal as possible we edited the XML so that they have the same operator weights and tuning values. Also, the population size used to generate the random starting tree differed so these were made the same as well.
The MCMC runs were run for 1 million steps in order to make them long enough that the slightly different ways extra likelihood calculations are done at the start for debugging purposes has little effect on the outcome. Also, with longer runs JIT compiler differences are eliminated. We took care to run the different programs under the same circumstances, on a computer not doing any other jobs at the time.
This whole process was automated to deal with the various data sets we wanted to test.
Threading
The way to set up threads in BEAST 2 is a bit cumbersome (v2.4.0 improves things a lot), so perhaps the reason is different configurations of threading. Therefore, we want to see what the impact of threading is. That led us to 3 variants:
- 1 thread BEAGLE SSE
- 2 thread BEAGLE SSE
- 4 thread BEAGLE SSE
For BEAST 1, we used the flags -overwrite -beagle_instances. For BEAST 2 we used -overwrite -threads for the SSE runs. For all cases, we verified that both programs use the same settings of BEAGLE as reported at the start of the run.
Data sets
To get an impression of the impact of different data, we randomly selected a number of data sets from treebase.org with a number of sizes. We also used the data sets from the BEAST 1 examples benchmark directory giving a total of 15 data sets.
| dataset |
taxa |
sites |
patterns |
| . |
. |
. |
. |
| M1044 |
50 |
1133 |
493 |
| M1366 |
41 |
1137 |
769 |
| M1510 |
36 |
1812 |
1020 |
| M1748 |
67 |
955 |
336 |
| M1749 |
74 |
2253 |
1673 |
| M1809 |
59 |
1824 |
1037 |
| M336 |
27 |
1949 |
934 |
| M3475 |
50 |
378 |
256 |
| M501 |
29 |
2520 |
1253 |
| M520 |
67 |
1098 |
534 |
| M755 |
64 |
1008 |
407 |
| M767 |
71 |
1082 |
446 |
| benchmark1 |
1441 |
98 |
593 |
| benchmark2 |
62 |
10869 |
5565 |
| old_benchmark |
17 |
1485 |
138 |
Versions
To have a fair comparison, we used the latest versions currently avaiable v1.8.3 and v2.4.0.
Results
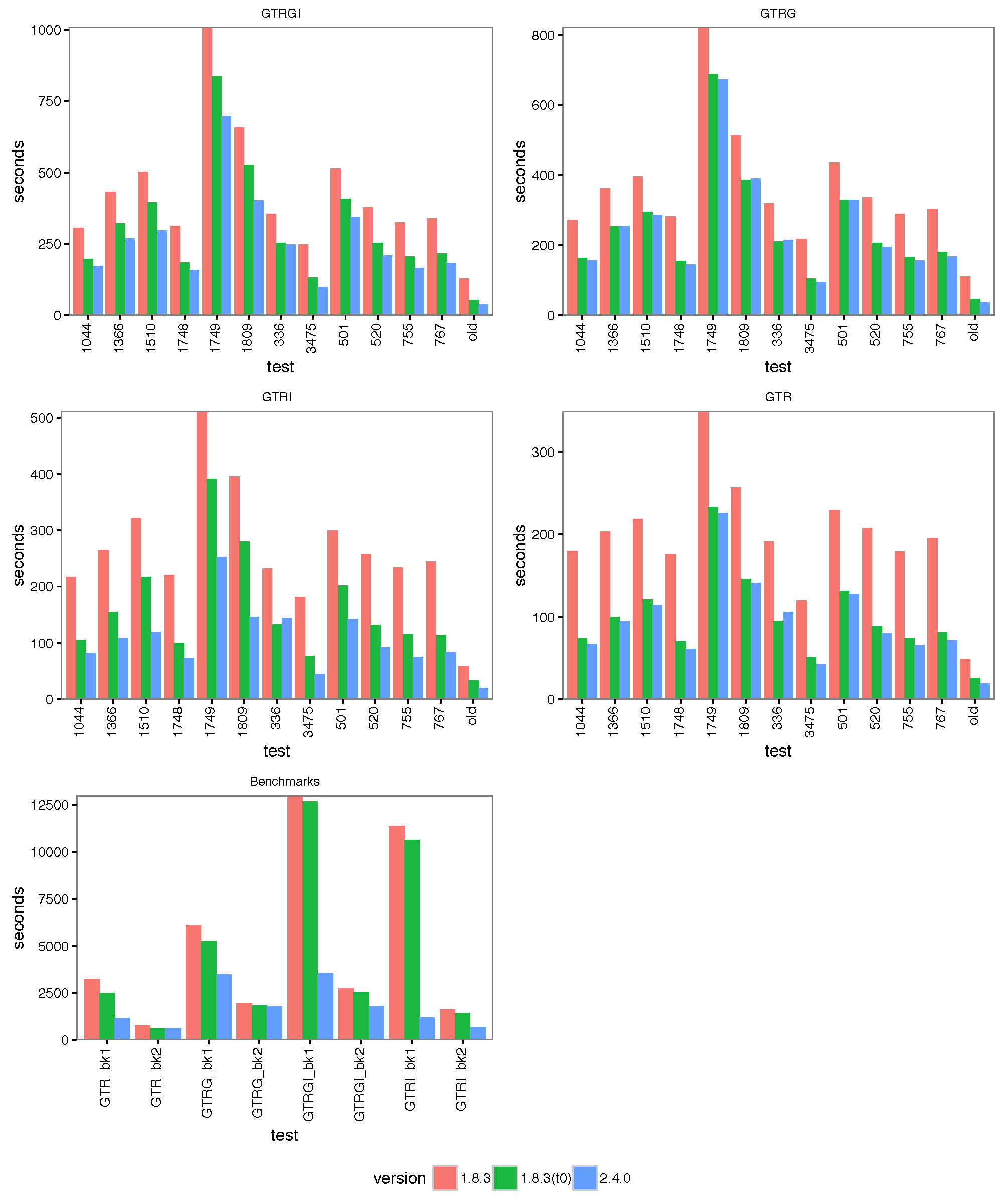
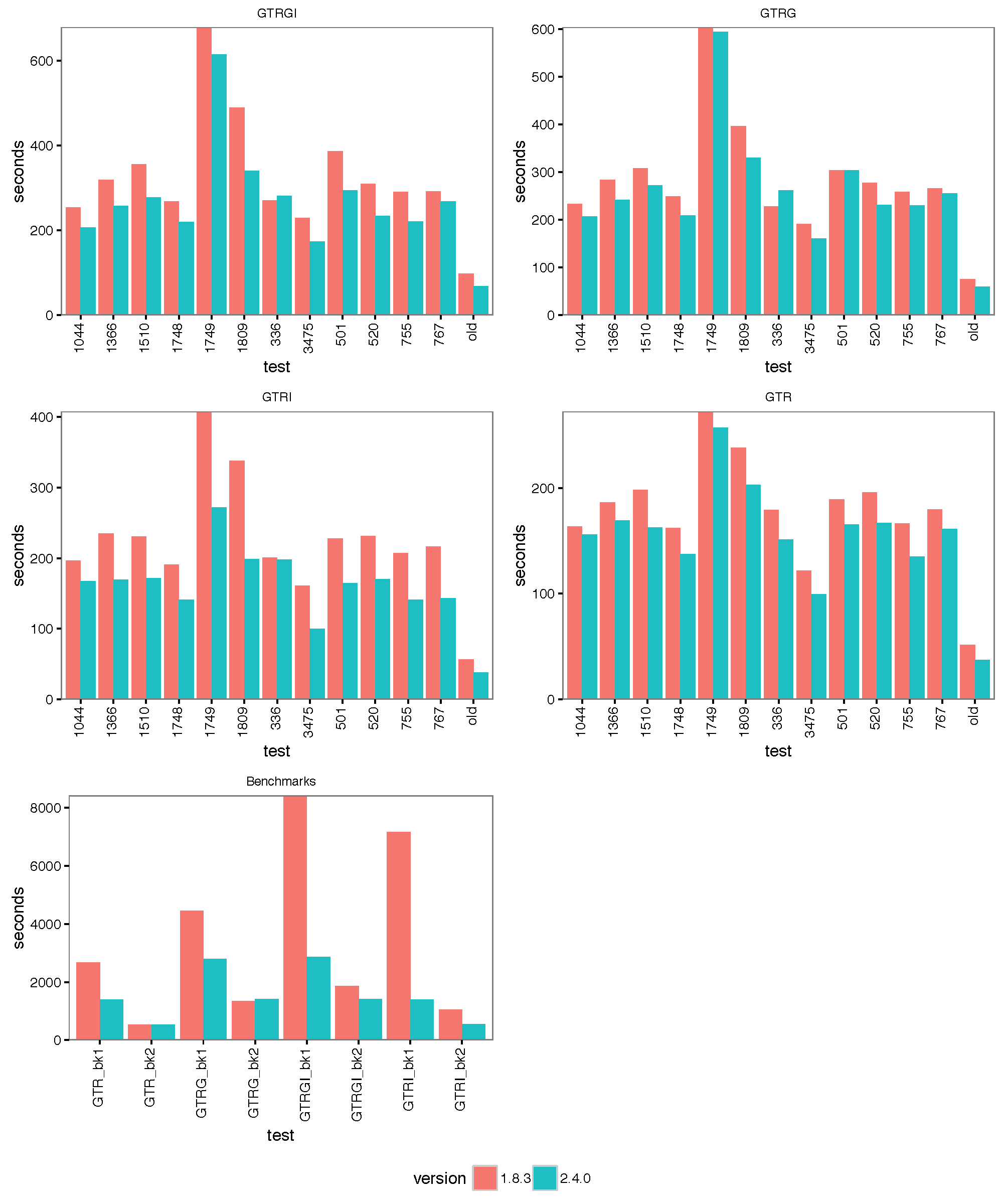
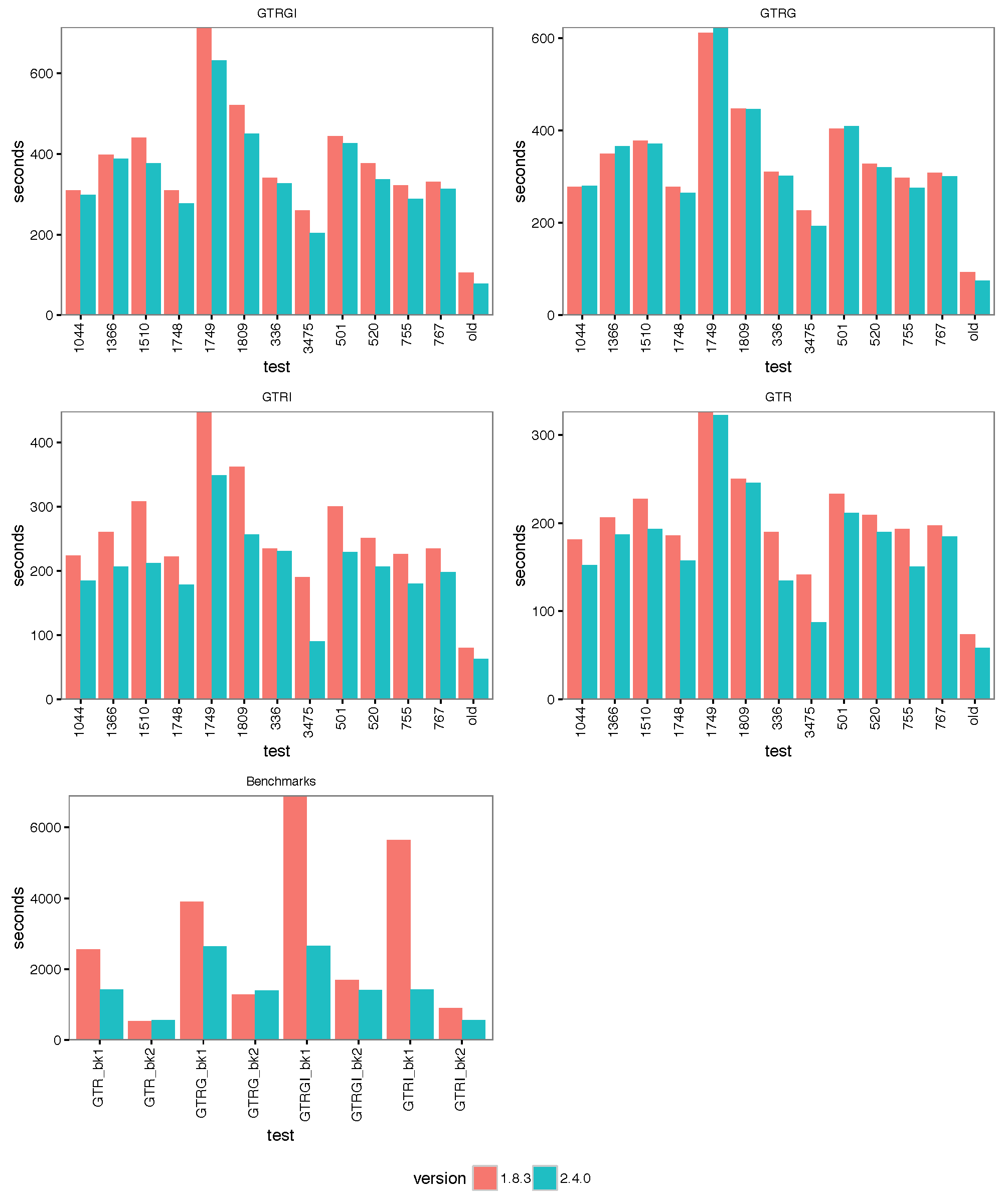
The images below show the run time for 1, 2, 4 thread(s) in Linux, where 1.8.3(t0) presents no threading pool for single thread in BEAST 1.8.3.
- 1 thread:
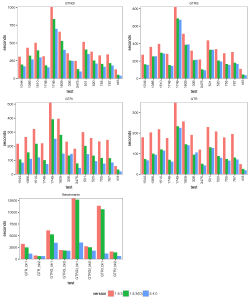
- 2 threads:
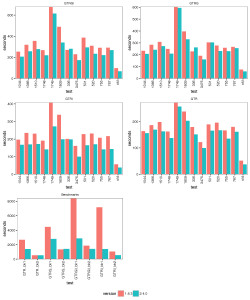
- 4 threads:

With increasing number of threads, the difference in run time in seconds decreases, but BEAST 2 is almost always slightly faster than BEAST 1 in these comparisons. However, it turned out that the data sets are too small for four threads to be of much use — the four threaded runs tended to be slower than for two threads, which is optimal for most of these datasets for both BEAST versions. This may also be a function of the hardware used.
Cursory checks of ESSs for BEAST 1 and 2 in Tracer did not show any substantial difference, which is not surprising since the same mixture of operators was used. Also, parameter estimates tended to agree between some randomly selected analyses.
To make sure that it differences are not OS dependent, we ran the analyses on Windows 7, OS X and Linux, but did not find any substantial differences between the operating systems.
Conclusions
To our surprise, we found that BEAST 2 is slightly faster than BEAST 1. This is not what we expected since both programs perform the same analysis using the same BEAGLE library. Although we did our best to compare apples with apples, it is possible we overlooked something, so let us know if you find anything that can explain the differences in performance.
If you want to replicate these runs, you can find them in the benchmark repository on https://github.com/CompEvol/benchmark, which includes the data, some instructions and scripts to run them.