BEAST improved performance
BEAST is up to 2x faster when using proportion invariant sites and BEAGLE. When using proportion invariant in combination with gamma rate heterogeneity, it still is faster than before.
BEAST always had a “beagle_instance” command line flag, that was essentially ignored. This is now replaced by an flag that actually works, and is named “instances” since it works both with java and BEAGLE tree likelihoods.
By default, the treelikelihood is now threaded for analyses using the Standard template in BEAUti. The number of threads used per treelikelihood is determined by the “instances” flag, but can be overridden using the “threads” attribute of ThreadedTreeLikelihood (which was migrated from the BEASTLabs package).
Further, there are a few minor performance improvements, including faster MRCAPrior handling.
A bug in StartBeastStartState was fixed to work with calibrations with other than the CalibratedYule prior.
BEAUti
The parametric distributions in priors panel now show mean as well as median of the distribution.
There is better taxon management preventing adding numbers to taxon names
The layout tip dates panel was improved to deal with changing window sizes.
A bug in *BEAST clock cloning is fixed.
Allow setting branch length as substitution option on tree logger, which was previously not possible.’
Improved JSON export of BEAST analyses (just use json as extension to the file when saving) and using a library with a more sensible license.
Package manager
The package manager has been changed so it can read all package information (including that of older versions) from a single package file. A bigger change is that BEAST is now treated as a separate package: when you start any of the BEAST applications, it loads the beast.jar file from the user package directory, and if it is not already there, will put a copy in that place. This makes it much easier to upgrade BEAST: just select BEAST in the package list and click the install/update button.
The GUI of the package manager is improved, among other things, showing by colour whether a package can be installed.
For developers
The biggest change with this release is really for developers, as outlined in a separate post here.
Packages
Due to some API changes, all packages have been re-released. Some packages have not been updated yet, but will be soon. New packages expected soon that have not been available before include startbeast2 and correlated characters.

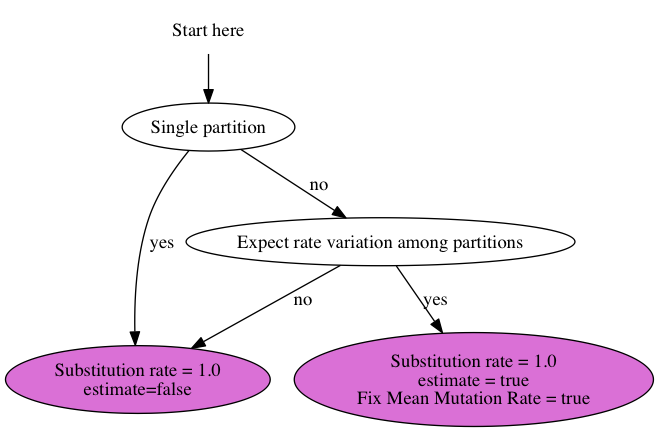
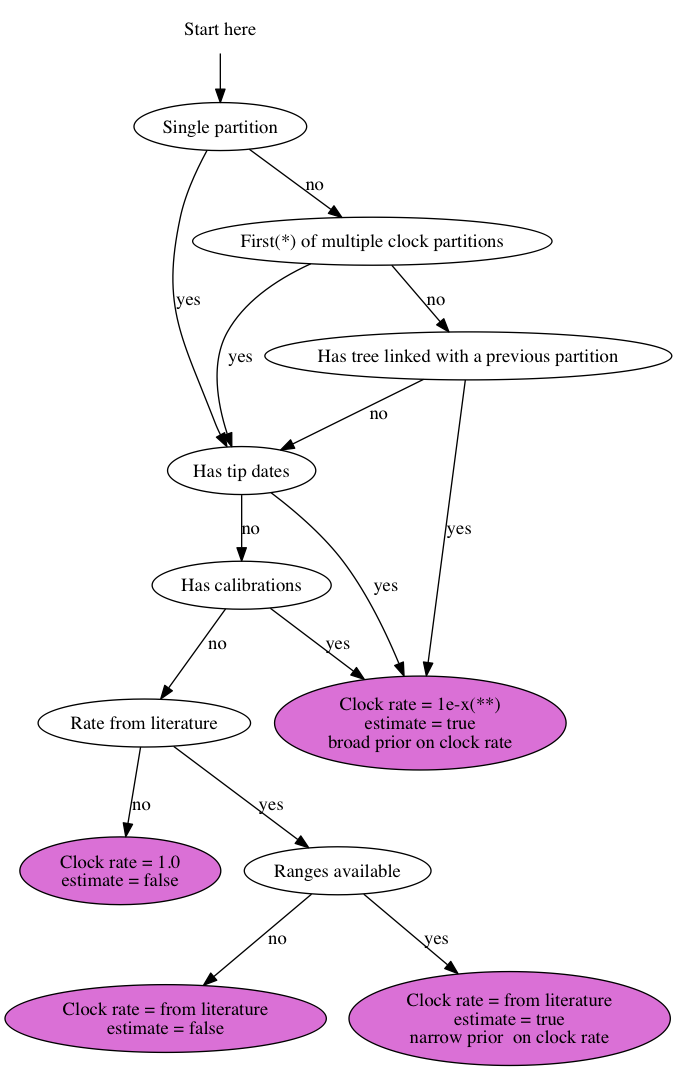
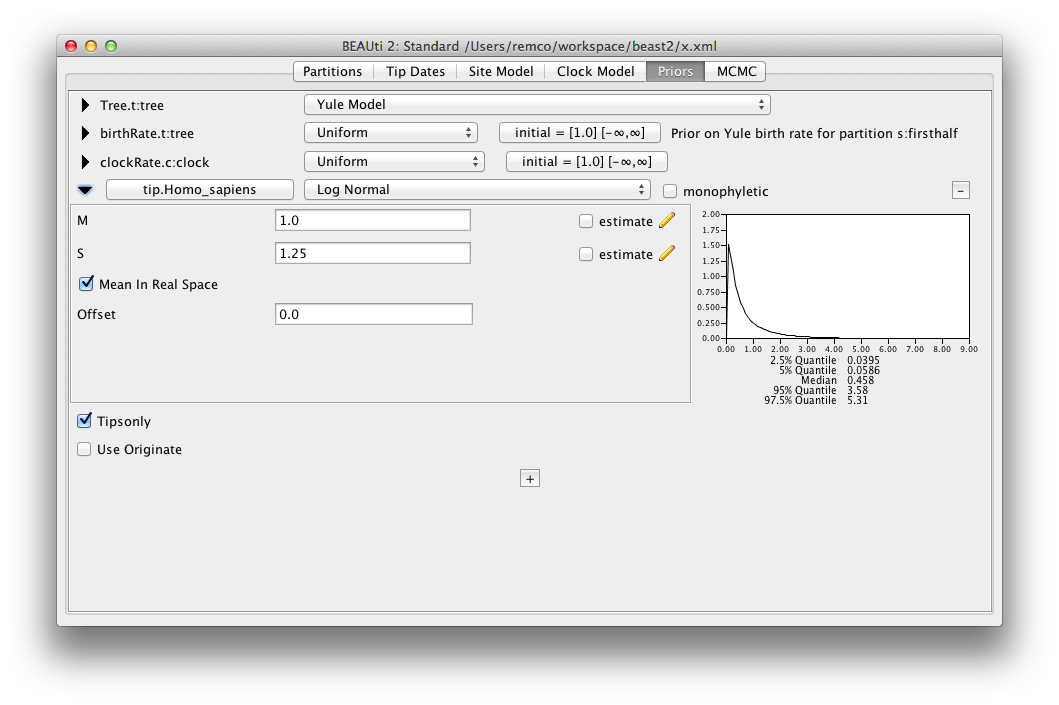
{kind=link}